

◆2024年度時実利彦記念賞受賞者 服部 信孝 先生 受賞の言葉
◆2024年度時実利彦記念賞受賞者 南部 篤 先生 受賞の言葉
◆2023年度塚原仲晃記念賞受賞者 西村 幸男 先生 受賞の言葉
◆2023年度塚原仲晃記念賞受賞者 岩崎 明子 先生 受賞の言葉
[会] 【追悼】ラリー・ヤング博士を偲んで
◆Awardee of 2024 Joseph Altman Award in Developmental Neuroscience, Shawn Sorrells
◆ブレイン・テック ガイドブック vol.2.0 −責任ある製品開発の手引き− パブリックコメント募集について(お願い)
◆2024年度 第26回時実利彦記念賞 受賞者決定のお知らせ
◆JNS-FENS 科学交流促進事業 FENS Forum 2024 参加支援Travel Awardの選考結果について
◆2024年度 JNS-CNS Exchange Travel Award Program 選考結果について
◆日本生物学オリンピック2024(熊本大会)のクラウドファンディングへの ご協力のお願い
◆2024年 第8回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
[会] 令和6年度 脳神経科学統合プログラム(個別重点研究課題) 公募開始のご案内
[会] 2024年 Neuroscience Research (NSR) 論文賞 受賞論文決定
◆The Brain Prize 2024 受賞者決定
[会] 第46回神経科学大会に関するChen Institute Science Writerによるレポート
[会] アンケート【他学会への入会状況と年次大会の合同開催に関する調査】 ご協力のお願い
◆THE BRAIN PRIZE 2024 WINNERS ANNOUNCED ON MARCH 5TH (CET)/ 6TH MARCH(JST)
[会] FENS Forum 2024 参加登録費会員価格特典のお知らせ
[会] 2023年度 臨時社員総会議事録
[会] 2024年度 学生会員/海外学生会員資格更新と卒業見込み時期登録のご案内
◆【応用脳科学コンソーシアムからのお知らせ】応用脳科学資格検定制度の創設について
[会] 第4回NBRP加齢マウス供給課題申請受付のご案内
[会] 2023 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 提言「未来の学術振興構想(2023年版)」の公表について
[会] 【新企画】「神経科学速報」スタートのお知らせ
[会] 【学会推薦枠】 2023年度 山田科学振興財団 研究援助 当学会会員が採択されました
◆FENS Summer School 2023 参加記 (Owusu Mensah Richard Nana Abankwah)
[会] 【応募期限延長】日本神経科学学会「ニューロナビゲータ2024」の募集について
◆Call for applications: Chief Editors of Neuroscience
◆【訃報】日本神経科学学会名誉会員 佐野 豊 先生
◆理事長からのメッセージ
◆2023年度 日本神経科学学会奨励賞受賞者 長谷川 恵美 先生
◆2023年度 日本神経科学学会奨励賞受賞者 乘本 裕明 先生
◆2023年度 日本神経科学学会奨励賞受賞者 田渕 理史 先生
◆2023年度 日本神経科学学会奨励賞受賞者 太田 茜 先生
◆2023年度 日本神経科学学会奨励賞受賞者 天羽 龍之介 先生
◆2022年度塚原仲晃記念賞受賞者 大木 研一 先生 受賞の言葉
[会] 研究活動をめぐる評価と研究公正に関する意識調査(webアンケート)ご協力のお願い
[会] 吉井聡先生のご逝去および「吉井聡君を送る会」の開催につきまして
◆Awardee of 2023 Joseph Altman Award in Developmental Neuroscience, Tomasz Nowakowski
◆2023年度時実利彦記念賞受賞者 坂野 仁 先生 受賞の言葉
◆2023年度時実利彦記念賞受賞者 今水 寛 先生 受賞の言葉
[会] 次期理事長選出のお知らせ
◆2023年度 第25回時実利彦記念賞 受賞者決定のお知らせ
◆2023年 Neuroscience Research (NSR) 論文賞 受賞論文決定
◆2023年 第7回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆The Brain Prize 2023 受賞者決定
[会] 【学会推薦枠】2023年度 ミズノスポーツ学等研究助成 当学会会員が採択されました
[会] 2023年度 学生会員/海外学生会員 更新手続きのご案内
[会] 『日本の大学・大学院における神経科学(脳科学)研究の倫理教育に関する調査』(Webアンケート)へのご協力のお願い
[会] 科学技術振興機構 ムーンショット型研究開発事業 プロジェクトマネージャー/課題 推進者の追加募集開始のお知らせ
◆評議員の名簿を公開しました
◆THE BRAIN PRIZE 2023 WINNERS ANNOUNCED ON 23RD MARCH(CET)/ 24TH MARCH(JST)
[会] 若手研究者の雇用に関するアンケート (男女共同参画学協会連絡会)の解析結果のご報告
[会] 【追悼】 小野武年先生を偲んで
[会] 【アンケート】科学技術系専門職における旧姓・通称使用に関する実態調査
[会] Neuroscience Research 誌の新編集体制
[会] Neuroscience Research 編集主幹を退任するにあたり
◆【訃報】日本神経科学学会名誉会員 小野 武年 先生
[会] 【学会推薦枠】 2022年度 日本学術振興会賞 当学会会員が受賞!(五十嵐啓 先生)
[会] 2022 総会報告
[会] SfN Neuroscience 2022 参加記
[会] SfN Neuroscience 2022 参加記
[会] 2023年度 理事選挙 結果公表
[会] JAXA 2022年度「きぼう」船内科学利用テーマの募集について
[会] 公開ワークショップ「Trusted Brain Tech / BMI の実現に向けて」へのご参加について(ご案内)
[会] 2023年度学生会員再登録期間変更のお知らせ
[会] 「新潟大学脳研究所年報2021」の発行について
[会] 国際基礎科学年(IYBSSD2022)YouTube プロモーションビデオの紹介
◆ニューロナビゲータ2023 のご紹介
[会] 2022年 FENS Forum 参加記
[会] コロナ禍におけるFENS Forum 2022
[会] 2022 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 【学会推薦枠】2022年度 山田科学振興財団 研究援助 当学会会員が採択されました
[会] 脳神経科学に関する意識調査(Webアンケート)へのご協力のお願い
[会] 「ブレイン・テック ガイドブック」パブリックコメント募集 について(お願い)
[会] BASENET* Hackathonのお知らせ
[会] コロナ禍における海外学術集会参加に関するネットワーク掲示板(日本分子生物学会HP)
[会] NEURO2022会期中の学会事務局について
◆日本神経科学学会「ニューロナビゲータ」の募集について
◆2022年度時実利彦記念賞受賞者 柳沢 正史 先生 受賞の言葉
[会] 若手研究者をとりまく評価に関する意識調査(Webアンケート)へのご協力のお願い
◆2022年度 日本神経科学学会奨励賞受賞者 柳下 祥 先生
◆2022年度 日本神経科学学会奨励賞受賞者 宮本 大祐 先生
◆2022年度 日本神経科学学会奨励賞受賞者 後藤 明弘 先生
◆2022年度 日本神経科学学会奨励賞受賞者 小野 大輔 先生
◆The Brain Prize ウェビナー – Circuit for Movement のお知らせ
[会] SfN北米神経科学学会 各種賞の募集のお知らせ
◆Awardee of 2022 Joseph Altman Award in Developmental Neuroscience, Denis Jabaudon
◆2022年度 JNS-CNS Exchange Travel Award Programの募集と選考結果について
◆2021年度塚原仲晃記念賞受賞者 南本 敬史 先生 受賞の言葉
◆2021年度塚原仲晃記念賞受賞者 松田 憲之 先生 受賞の言葉
◆2022年度時実利彦記念賞受賞者 長谷川 成人 先生 受賞の言葉
[会] The Brain Prize 2023 受賞候補者 - 推薦募集
◆2022年度 第22回日本神経科学学会奨励賞 受賞者決定!
[会] 若手研究者の雇用に関するアンケート(男女共同参画学協会連絡会)
◆2022年度 第24回時実利彦記念賞 受賞者決定のお知らせ
[会] 追悼コーナー「時代の肖像~時代を築いた先人へ捧げるメッセージ」開設のお知らせ
◆2022年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆公益財団法人ブレインサイエンス振興財団 2021年度 塚原仲晃記念賞及び研究助成受領者
[会] SfN:Peter Seeburg Prize- Nominations Closing Soon
◆The Brain Prize 2022 受賞者決定
[会] FENS Forum 2022 参加登録費会員価格特典のお知らせ
◆2022年 Neuroscience Research (NSR) 論文賞 受賞論文決定
[会] 【学会推薦枠】第62回(2021年度)東レ科学技術研究助成 当学会会員2名が受領者に決定!
◆THE BRAIN PRIZE 2022 WINNERS ANNOUNCED ON 3RD MARCH(CET)/ 4TH MARCH(JST)
◆JNS-FENS 科学交流促進事業 FENS Forum 2022 参加支援Travel Award募集と選考結果について
[会] 2021年 Cajal Course参加記
◆SfN2022セッション企画提案の募集のお知らせ
◆Declaration on Research Assessmentへの署名について
◆【学会推薦枠】第4回(2021年度)島津奨励賞 当学会会員が受賞!(村山正宜 先生)
[会] Neuroscience Research (NSR) 編集主幹公募のお知らせ
[会] FENS Forum 2022 参加登録費会員価格特典のお知らせ
◆霊長類研究所の改編の方向性について
◆Virtual SfN 2021: Global Neuroscience Social のご案内
[会] 2022年 学生会員の再登録と若手会員制度のご案内
◆【ご協力のお願い】科学技術系専門職の男女共同参画実態調査(大規模アンケート)
[会] 2021 総会報告
◆2021年度 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 第43回(2020年)日本神経科学大会 演題抄録閲覧時のパスワードについて
◆大学院生を対象にしたアンケート調査(2021年)
◆【訃報】日本神経科学学会名誉会員 小幡 邦彦 先生
[会] 日本神経科学学会 法人化のお知らせ
◆2021年度 日本神経科学学会奨励賞受賞者 Dr. Aurelio Cortese
◆2021年度 日本神経科学学会奨励賞受賞者 髙野 哲也 先生
◆2021年度 日本神経科学学会奨励賞受賞者 山口 隆司 先生
◆2021年度 日本神経科学学会奨励賞受賞者 岩田 亮平 先生
◆2021年度 日本神経科学学会奨励賞受賞者 井上 清香 先生
◆日本神経科学学会 機関誌 Neuroscience Research 論文賞のご案内
◆Awardee of 2021 Joseph Altman Award in Developmental Neuroscience, Sergiu P. Pasca
◆NPO法人「脳の世紀推進会議」の紹介と入会のお願い
◆2020年度塚原仲晃記念賞受賞者 松崎 政紀 先生
◆2020年度塚原仲晃記念賞受賞者 古川 浩康 先生
◆2021年度時実利彦記念賞受賞者 野田 昌晴 先生
[会] 「霊長類を対象とする実験ガイドライン」一部改訂のお知らせ
◆2021年度 第23回時実利彦記念賞 受賞者決定
◆公益財団法人ブレインサイエンス振興財団 2020年度 塚原仲晃記念賞及び研究助成受領者
◆2021年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆文科省の令和3年度の戦略的創造研究推進事業の戦略目標等の決定について
◆学会ホームページがスマートフォン対応になりました
◆2021年The Brain Prize受賞者が決定しました!
◆会長からのメッセージ
◆【JST未来社会創造事業(探索加速型)】 重点公募テーマ策定のためのアイデア募集開始のお知らせ
◆日本神経科学学会はALBA Declarationを支持しています
◆第5次男女共同参画基本計画が閣議決定されました
[会] 2021年学生会員の再登録と若手会員登録のご案内
[会] 会員サイトリニューアルのお知らせ 【会員情報確認のお願い】
[会] 緊急声明の表出について
[会] 2020 総会報告
◆綜合画像研究支援より日本人研究者・技術者の国際動向調査のお願い
◆2020年度 国際宇宙ステーション(ISS)・「きぼう」利用テーマ募集のお知らせ
[会] 神経科学分野における霊長類を対象とする実験ガイドライン
◆Call for Papers for a Special Issue of Brain Structure and Function
◆緊急事態宣言による在宅勤務中の科学者・技術者の実態調査結果報告
◆日本脳科学関連学会連合よりの緊急提言について
◆成体脳における自然/人為的なニューロン新生
◆細胞の個性に応じた嗅神経回路形成の分子的基盤
◆海馬において空間および文脈記憶を司る二つの記憶痕跡の形成および作用機序の解明
◆個体脳の情報処理を細胞生理学的現象として理解する
◆神経情報処理のロジックを解明する
◆2019年度塚原仲晃記念賞受賞者 高橋 琢哉 先生
◆大学院生を対象にしたアンケート調査
[会] 緊急事態宣言による在宅勤務中の科学者・技術者の実態調査ご協力のお願い
◆生物科学学会連合より緊急声明(2) ~緊急事態措置による影響緩和のための各関係機関へのお願い~
◆2020年度時実利彦記念賞受賞者 尾崎 紀夫 先生
◆2020年度時実利彦記念賞受賞者 鍋倉 淳一 先生
◆2020年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆Awardee of 2020 Joseph Altman Award in Developmental Neuroscience, Haruki Takeuchi
◆遺伝研 新型コロナウイルスによる緊急事態対応事業
◆日本アルコール・アディクション医学会から注意喚起のお願い 新型コロナウイルス問題で心配される アルコール依存症やゲーム障害等のアディクション
◆新型コロナウイルス感染拡大に際して
◆公益財団法人ブレインサイエンス振興財団 2019年度 塚原仲晃記念賞及び研究助成受領者決定
◆2020年度 第22回時実利彦記念賞 受賞者決定
◆「科学コミュニケーションガイドライン」を公開しました
◆国際生物学オリンピック(IBO2020)ふるさと納税での寄付のご案内
◆2019年度 JNS-SfN Exchange Travel Awardの募集と選考結果について
◆◆「遺伝資源の研究開発と提供国措置の影響評価に関するアンケート」 ご協力のお願い◆
◆FENS Forum 2020 参加登録費会員価格特典のお知らせ
[会] 2020年学生会員の再登録と若手会員登録のご案内
◆【訃報】日本神経科学学会名誉会員 濱清先生
◆動物実験委員会から署名のお願い
◆日本神経科学学会パネル理事選挙 電子投票のお知らせ
◆日本神経科学学会マスコットキャラクター決定のお知らせ
◆[日本心理学会] Japanese Psychological Research特集号への投稿のお願い
◆NEURO2019での学会デスク設置のご案内と事務局休業のお知らせ
◆ランチョン大討論会 「次の20年にどうやって脳科学にブレークスルーを生むか?」
◆2019年度 第19回日本神経科学学会奨励賞 受賞者決定
◆脳の機能を分子レベルで理解する
◆海馬における自己の場所と他者の場所の表象
◆固有知覚による運動制御と運動機能回復のメカニズム
◆脳発達から人間性の起原を探る
◆神経回路形成・再編成における細胞骨格セプチンの役割
◆2019年度時実利彦記念賞受賞者 林 康紀 先生
◆【脳科連】AMED国際脳事業およびInternational Brain Initiative ご案内
◆公示「日本神経科学学会理事選挙について」
◆第33回塚原仲晃記念賞受賞者 Thomas McHugh先生
◆神経科学ニュースを新デザインの封筒でお届けします
◆Altman Award in Developmental Neuroscience
◆公益財団法人ブレインサイエンス振興財団 平成30年度 塚原仲晃記念賞及び研究助成受領者決定
◆2019年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆「第11回科学技術予測調査」へのご協力のお願い (文部科学省科学技術・学術政策研究所)
◆2019年度 第21回時実利彦記念賞 受賞者決定
◆【訃報】日本神経科学学会名誉会員 島津浩 先生
◆日本神経科学学会 マスコットキャラクター投票締め切り迫る!
◆2019 ANS Conference, Australasian Neuroscience Society – 39th Annual Scientific Meeting 参加登録費会員価格特典のお知らせ
◆ニューロン樹状突起で生じる知覚スイッチ
◆認知症問題の解決に向けた神経科学からのアプローチ
◆脳が学習によって変化する様子
◆遺伝子導入、編集、標識技術による単一神経細胞内の分子イメージング
◆前頭前皮質内局所回路における作業記憶の制御機構
◆「平成30年度 時実利彦記念賞受賞者 小松 英彦 先生」
◆第32回塚原仲晃記念賞受賞者 内田 直滋先生
◆Altman Award in Developmental Neuroscience
◆平成30年度ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆Participating report of the 12th biennial Conference of CNS : Kai Li
◆CAJAL Course参加記
◆Cajal Advanced Neuroscience Training Programme参加記
◆Participating report of the 12th biennial Conference of CNS : Kelvin Hui
◆「平成29年度 時実利彦記念賞受賞者 齊藤 実先生」
◆「第1回 ジョセフ・アルトマン記念発達神経科学賞受賞者 今吉 格先生」
◆第31回塚原仲晃記念賞受賞者 磯田 昌岐先生
◆第31回塚原仲晃記念賞受賞者 安田 涼平先生
◆小さな脳から探る神経回路
◆大人の脳を維持する仕組み
◆「遺伝学的及び光学的手法による脳内神経活動の可視化と機能解析への応用」
◆「社会」を紐解くニューロサイエンス
◆「海馬への異なる脳領域からの入力の機能と制御」
◆平成29年度 第1回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆第30回塚原仲晃記念賞受賞者 樋口真人先生
◆第30回塚原仲晃記念賞受賞者 坂場武史先生
◆平成28年度時実利彦記念賞受賞者 影山龍一郎先生「神経幹細胞の増殖と分化制御機構の解明とその操作」
◆小型魚類を用いた神経精神疾患研究
◆嗅覚情報処理を支える神経回路の形成基盤
◆視覚から行動へ
◆状況に応じた柔軟な情報処理を可能にする脳神経回路
◆「脳とは何か」の答えを目指して
◆光の動きを検知する視覚神経回路の機能と発達
◆精神疾患病態としてのシナプス病理解明とその治療戦略への展開
◆意識経験はどのような神経メカニズムによって支えられているのか?
◆記憶形成・記憶保持を負に制御する神経回路の研究
◆脳の設計図と変化する脳
◆平成27年度時実利彦記念賞受賞者 渡辺雅彦先生「神経活動依存的な神経回路発達と回路機能発現に関する分子解剖学的研究」
◆第29回塚原仲晃記念賞受賞者 池谷 裕二先生 脳回路活動の構造解析」
◆時差消失マウスの開発による時差の神経分子シグナルの研究
◆サル側頭葉における物体間対連合の視覚表象及び記憶想起を司る神経回路の計算原理
◆てんかんの細胞神経科学
◆嗅覚から記憶へ
◆神経細胞の分化と可塑性を制御するRNAプログラム
◆第28回塚原仲晃記念賞受賞者榎本 和生先生「感覚ニューロン受容野の自己組織化と再編機構の解明」
◆平成26年度時実利彦記念賞受賞者岡本仁先生「脊椎動物脳の進化的保存を利用した情動制御機構の解明」
◆第28回塚原仲晃記念賞受賞者加藤忠史先生 精神疾患の神経生物学的研究」
◆精神医学領域のUnmet medical needsの解消を目指して
◆単純な学習系で照らし出す大脳新皮質に記憶が固定化される仕組み
◆中枢神経疾患における組織傷害と修復を制御する生体応答
◆分子1個1個のふるまいから脳細胞の自己組織化機構を読み解く
◆小脳におけるシナプス形成機構の解明
◆軸索における活動電位の伝導様式とグリア細胞の関与
◆予想に反した結果が発見につながったとき
◆線虫をもちいた温度応答の分子神経メカニズムの解析
◆地図を手に山を登る
◆精神・神経疾患の社会性障害の理解と克服に向けて
◆体液Naレベルのセンシング〜グリア細胞による ニューロンの制御、そして疾患
◆神経科学と心理学の橋渡しを目指して
◆好奇心の原点へ
◆大脳基底核神経回路の制御機構
◆神経軸索ガイダンスの駆動メカニズムの解明を目指して
◆2024年度時実利彦記念賞受賞者 南部 篤 先生 受賞の言葉
◆2023年度塚原仲晃記念賞受賞者 西村 幸男 先生 受賞の言葉
◆2023年度塚原仲晃記念賞受賞者 岩崎 明子 先生 受賞の言葉
[会] 【追悼】ラリー・ヤング博士を偲んで
◆Awardee of 2024 Joseph Altman Award in Developmental Neuroscience, Shawn Sorrells
◆ブレイン・テック ガイドブック vol.2.0 −責任ある製品開発の手引き− パブリックコメント募集について(お願い)
◆2024年度 第26回時実利彦記念賞 受賞者決定のお知らせ
◆JNS-FENS 科学交流促進事業 FENS Forum 2024 参加支援Travel Awardの選考結果について
◆2024年度 JNS-CNS Exchange Travel Award Program 選考結果について
◆日本生物学オリンピック2024(熊本大会)のクラウドファンディングへの ご協力のお願い
◆2024年 第8回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
[会] 令和6年度 脳神経科学統合プログラム(個別重点研究課題) 公募開始のご案内
[会] 2024年 Neuroscience Research (NSR) 論文賞 受賞論文決定
◆The Brain Prize 2024 受賞者決定
[会] 第46回神経科学大会に関するChen Institute Science Writerによるレポート
[会] アンケート【他学会への入会状況と年次大会の合同開催に関する調査】 ご協力のお願い
◆THE BRAIN PRIZE 2024 WINNERS ANNOUNCED ON MARCH 5TH (CET)/ 6TH MARCH(JST)
[会] FENS Forum 2024 参加登録費会員価格特典のお知らせ
[会] 2023年度 臨時社員総会議事録
[会] 2024年度 学生会員/海外学生会員資格更新と卒業見込み時期登録のご案内
◆【応用脳科学コンソーシアムからのお知らせ】応用脳科学資格検定制度の創設について
[会] 第4回NBRP加齢マウス供給課題申請受付のご案内
[会] 2023 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 提言「未来の学術振興構想(2023年版)」の公表について
[会] 【新企画】「神経科学速報」スタートのお知らせ
[会] 【学会推薦枠】 2023年度 山田科学振興財団 研究援助 当学会会員が採択されました
◆FENS Summer School 2023 参加記 (Owusu Mensah Richard Nana Abankwah)
[会] 【応募期限延長】日本神経科学学会「ニューロナビゲータ2024」の募集について
◆Call for applications: Chief Editors of Neuroscience
◆【訃報】日本神経科学学会名誉会員 佐野 豊 先生
◆理事長からのメッセージ
◆2023年度 日本神経科学学会奨励賞受賞者 長谷川 恵美 先生
◆2023年度 日本神経科学学会奨励賞受賞者 乘本 裕明 先生
◆2023年度 日本神経科学学会奨励賞受賞者 田渕 理史 先生
◆2023年度 日本神経科学学会奨励賞受賞者 太田 茜 先生
◆2023年度 日本神経科学学会奨励賞受賞者 天羽 龍之介 先生
◆2022年度塚原仲晃記念賞受賞者 大木 研一 先生 受賞の言葉
[会] 研究活動をめぐる評価と研究公正に関する意識調査(webアンケート)ご協力のお願い
[会] 吉井聡先生のご逝去および「吉井聡君を送る会」の開催につきまして
◆Awardee of 2023 Joseph Altman Award in Developmental Neuroscience, Tomasz Nowakowski
◆2023年度時実利彦記念賞受賞者 坂野 仁 先生 受賞の言葉
◆2023年度時実利彦記念賞受賞者 今水 寛 先生 受賞の言葉
[会] 次期理事長選出のお知らせ
◆2023年度 第25回時実利彦記念賞 受賞者決定のお知らせ
◆2023年 Neuroscience Research (NSR) 論文賞 受賞論文決定
◆2023年 第7回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆The Brain Prize 2023 受賞者決定
[会] 【学会推薦枠】2023年度 ミズノスポーツ学等研究助成 当学会会員が採択されました
[会] 2023年度 学生会員/海外学生会員 更新手続きのご案内
[会] 『日本の大学・大学院における神経科学(脳科学)研究の倫理教育に関する調査』(Webアンケート)へのご協力のお願い
[会] 科学技術振興機構 ムーンショット型研究開発事業 プロジェクトマネージャー/課題 推進者の追加募集開始のお知らせ
◆評議員の名簿を公開しました
◆THE BRAIN PRIZE 2023 WINNERS ANNOUNCED ON 23RD MARCH(CET)/ 24TH MARCH(JST)
[会] 若手研究者の雇用に関するアンケート (男女共同参画学協会連絡会)の解析結果のご報告
[会] 【追悼】 小野武年先生を偲んで
[会] 【アンケート】科学技術系専門職における旧姓・通称使用に関する実態調査
[会] Neuroscience Research 誌の新編集体制
[会] Neuroscience Research 編集主幹を退任するにあたり
◆【訃報】日本神経科学学会名誉会員 小野 武年 先生
[会] 【学会推薦枠】 2022年度 日本学術振興会賞 当学会会員が受賞!(五十嵐啓 先生)
[会] 2022 総会報告
[会] SfN Neuroscience 2022 参加記
[会] SfN Neuroscience 2022 参加記
[会] 2023年度 理事選挙 結果公表
[会] JAXA 2022年度「きぼう」船内科学利用テーマの募集について
[会] 公開ワークショップ「Trusted Brain Tech / BMI の実現に向けて」へのご参加について(ご案内)
[会] 2023年度学生会員再登録期間変更のお知らせ
[会] 「新潟大学脳研究所年報2021」の発行について
[会] 国際基礎科学年(IYBSSD2022)YouTube プロモーションビデオの紹介
◆ニューロナビゲータ2023 のご紹介
[会] 2022年 FENS Forum 参加記
[会] コロナ禍におけるFENS Forum 2022
[会] 2022 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 【学会推薦枠】2022年度 山田科学振興財団 研究援助 当学会会員が採択されました
[会] 脳神経科学に関する意識調査(Webアンケート)へのご協力のお願い
[会] 「ブレイン・テック ガイドブック」パブリックコメント募集 について(お願い)
[会] BASENET* Hackathonのお知らせ
[会] コロナ禍における海外学術集会参加に関するネットワーク掲示板(日本分子生物学会HP)
[会] NEURO2022会期中の学会事務局について
◆日本神経科学学会「ニューロナビゲータ」の募集について
◆2022年度時実利彦記念賞受賞者 柳沢 正史 先生 受賞の言葉
[会] 若手研究者をとりまく評価に関する意識調査(Webアンケート)へのご協力のお願い
◆2022年度 日本神経科学学会奨励賞受賞者 柳下 祥 先生
◆2022年度 日本神経科学学会奨励賞受賞者 宮本 大祐 先生
◆2022年度 日本神経科学学会奨励賞受賞者 後藤 明弘 先生
◆2022年度 日本神経科学学会奨励賞受賞者 小野 大輔 先生
◆The Brain Prize ウェビナー – Circuit for Movement のお知らせ
[会] SfN北米神経科学学会 各種賞の募集のお知らせ
◆Awardee of 2022 Joseph Altman Award in Developmental Neuroscience, Denis Jabaudon
◆2022年度 JNS-CNS Exchange Travel Award Programの募集と選考結果について
◆2021年度塚原仲晃記念賞受賞者 南本 敬史 先生 受賞の言葉
◆2021年度塚原仲晃記念賞受賞者 松田 憲之 先生 受賞の言葉
◆2022年度時実利彦記念賞受賞者 長谷川 成人 先生 受賞の言葉
[会] The Brain Prize 2023 受賞候補者 - 推薦募集
◆2022年度 第22回日本神経科学学会奨励賞 受賞者決定!
[会] 若手研究者の雇用に関するアンケート(男女共同参画学協会連絡会)
◆2022年度 第24回時実利彦記念賞 受賞者決定のお知らせ
[会] 追悼コーナー「時代の肖像~時代を築いた先人へ捧げるメッセージ」開設のお知らせ
◆2022年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆公益財団法人ブレインサイエンス振興財団 2021年度 塚原仲晃記念賞及び研究助成受領者
[会] SfN:Peter Seeburg Prize- Nominations Closing Soon
◆The Brain Prize 2022 受賞者決定
[会] FENS Forum 2022 参加登録費会員価格特典のお知らせ
◆2022年 Neuroscience Research (NSR) 論文賞 受賞論文決定
[会] 【学会推薦枠】第62回(2021年度)東レ科学技術研究助成 当学会会員2名が受領者に決定!
◆THE BRAIN PRIZE 2022 WINNERS ANNOUNCED ON 3RD MARCH(CET)/ 4TH MARCH(JST)
◆JNS-FENS 科学交流促進事業 FENS Forum 2022 参加支援Travel Award募集と選考結果について
[会] 2021年 Cajal Course参加記
◆SfN2022セッション企画提案の募集のお知らせ
◆Declaration on Research Assessmentへの署名について
◆【学会推薦枠】第4回(2021年度)島津奨励賞 当学会会員が受賞!(村山正宜 先生)
[会] Neuroscience Research (NSR) 編集主幹公募のお知らせ
[会] FENS Forum 2022 参加登録費会員価格特典のお知らせ
◆霊長類研究所の改編の方向性について
◆Virtual SfN 2021: Global Neuroscience Social のご案内
[会] 2022年 学生会員の再登録と若手会員制度のご案内
◆【ご協力のお願い】科学技術系専門職の男女共同参画実態調査(大規模アンケート)
[会] 2021 総会報告
◆2021年度 JNS-SfN Exchange Travel Awardの募集と選考結果について
[会] 第43回(2020年)日本神経科学大会 演題抄録閲覧時のパスワードについて
◆大学院生を対象にしたアンケート調査(2021年)
◆【訃報】日本神経科学学会名誉会員 小幡 邦彦 先生
[会] 日本神経科学学会 法人化のお知らせ
◆2021年度 日本神経科学学会奨励賞受賞者 Dr. Aurelio Cortese
◆2021年度 日本神経科学学会奨励賞受賞者 髙野 哲也 先生
◆2021年度 日本神経科学学会奨励賞受賞者 山口 隆司 先生
◆2021年度 日本神経科学学会奨励賞受賞者 岩田 亮平 先生
◆2021年度 日本神経科学学会奨励賞受賞者 井上 清香 先生
◆日本神経科学学会 機関誌 Neuroscience Research 論文賞のご案内
◆Awardee of 2021 Joseph Altman Award in Developmental Neuroscience, Sergiu P. Pasca
◆NPO法人「脳の世紀推進会議」の紹介と入会のお願い
◆2020年度塚原仲晃記念賞受賞者 松崎 政紀 先生
◆2020年度塚原仲晃記念賞受賞者 古川 浩康 先生
◆2021年度時実利彦記念賞受賞者 野田 昌晴 先生
[会] 「霊長類を対象とする実験ガイドライン」一部改訂のお知らせ
◆2021年度 第23回時実利彦記念賞 受賞者決定
◆公益財団法人ブレインサイエンス振興財団 2020年度 塚原仲晃記念賞及び研究助成受領者
◆2021年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆文科省の令和3年度の戦略的創造研究推進事業の戦略目標等の決定について
◆学会ホームページがスマートフォン対応になりました
◆2021年The Brain Prize受賞者が決定しました!
◆会長からのメッセージ
◆【JST未来社会創造事業(探索加速型)】 重点公募テーマ策定のためのアイデア募集開始のお知らせ
◆日本神経科学学会はALBA Declarationを支持しています
◆第5次男女共同参画基本計画が閣議決定されました
[会] 2021年学生会員の再登録と若手会員登録のご案内
[会] 会員サイトリニューアルのお知らせ 【会員情報確認のお願い】
[会] 緊急声明の表出について
[会] 2020 総会報告
◆綜合画像研究支援より日本人研究者・技術者の国際動向調査のお願い
◆2020年度 国際宇宙ステーション(ISS)・「きぼう」利用テーマ募集のお知らせ
[会] 神経科学分野における霊長類を対象とする実験ガイドライン
◆Call for Papers for a Special Issue of Brain Structure and Function
◆緊急事態宣言による在宅勤務中の科学者・技術者の実態調査結果報告
◆日本脳科学関連学会連合よりの緊急提言について
◆成体脳における自然/人為的なニューロン新生
◆細胞の個性に応じた嗅神経回路形成の分子的基盤
◆海馬において空間および文脈記憶を司る二つの記憶痕跡の形成および作用機序の解明
◆個体脳の情報処理を細胞生理学的現象として理解する
◆神経情報処理のロジックを解明する
◆2019年度塚原仲晃記念賞受賞者 高橋 琢哉 先生
◆大学院生を対象にしたアンケート調査
[会] 緊急事態宣言による在宅勤務中の科学者・技術者の実態調査ご協力のお願い
◆生物科学学会連合より緊急声明(2) ~緊急事態措置による影響緩和のための各関係機関へのお願い~
◆2020年度時実利彦記念賞受賞者 尾崎 紀夫 先生
◆2020年度時実利彦記念賞受賞者 鍋倉 淳一 先生
◆2020年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆Awardee of 2020 Joseph Altman Award in Developmental Neuroscience, Haruki Takeuchi
◆遺伝研 新型コロナウイルスによる緊急事態対応事業
◆日本アルコール・アディクション医学会から注意喚起のお願い 新型コロナウイルス問題で心配される アルコール依存症やゲーム障害等のアディクション
◆新型コロナウイルス感染拡大に際して
◆公益財団法人ブレインサイエンス振興財団 2019年度 塚原仲晃記念賞及び研究助成受領者決定
◆2020年度 第22回時実利彦記念賞 受賞者決定
◆「科学コミュニケーションガイドライン」を公開しました
◆国際生物学オリンピック(IBO2020)ふるさと納税での寄付のご案内
◆2019年度 JNS-SfN Exchange Travel Awardの募集と選考結果について
◆◆「遺伝資源の研究開発と提供国措置の影響評価に関するアンケート」 ご協力のお願い◆
◆FENS Forum 2020 参加登録費会員価格特典のお知らせ
[会] 2020年学生会員の再登録と若手会員登録のご案内
◆【訃報】日本神経科学学会名誉会員 濱清先生
◆動物実験委員会から署名のお願い
◆日本神経科学学会パネル理事選挙 電子投票のお知らせ
◆日本神経科学学会マスコットキャラクター決定のお知らせ
◆[日本心理学会] Japanese Psychological Research特集号への投稿のお願い
◆NEURO2019での学会デスク設置のご案内と事務局休業のお知らせ
◆ランチョン大討論会 「次の20年にどうやって脳科学にブレークスルーを生むか?」
◆2019年度 第19回日本神経科学学会奨励賞 受賞者決定
◆脳の機能を分子レベルで理解する
◆海馬における自己の場所と他者の場所の表象
◆固有知覚による運動制御と運動機能回復のメカニズム
◆脳発達から人間性の起原を探る
◆神経回路形成・再編成における細胞骨格セプチンの役割
◆2019年度時実利彦記念賞受賞者 林 康紀 先生
◆【脳科連】AMED国際脳事業およびInternational Brain Initiative ご案内
◆公示「日本神経科学学会理事選挙について」
◆第33回塚原仲晃記念賞受賞者 Thomas McHugh先生
◆神経科学ニュースを新デザインの封筒でお届けします
◆Altman Award in Developmental Neuroscience
◆公益財団法人ブレインサイエンス振興財団 平成30年度 塚原仲晃記念賞及び研究助成受領者決定
◆2019年ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆「第11回科学技術予測調査」へのご協力のお願い (文部科学省科学技術・学術政策研究所)
◆2019年度 第21回時実利彦記念賞 受賞者決定
◆【訃報】日本神経科学学会名誉会員 島津浩 先生
◆日本神経科学学会 マスコットキャラクター投票締め切り迫る!
◆2019 ANS Conference, Australasian Neuroscience Society – 39th Annual Scientific Meeting 参加登録費会員価格特典のお知らせ
◆ニューロン樹状突起で生じる知覚スイッチ
◆認知症問題の解決に向けた神経科学からのアプローチ
◆脳が学習によって変化する様子
◆遺伝子導入、編集、標識技術による単一神経細胞内の分子イメージング
◆前頭前皮質内局所回路における作業記憶の制御機構
◆「平成30年度 時実利彦記念賞受賞者 小松 英彦 先生」
◆第32回塚原仲晃記念賞受賞者 内田 直滋先生
◆Altman Award in Developmental Neuroscience
◆平成30年度ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆Participating report of the 12th biennial Conference of CNS : Kai Li
◆CAJAL Course参加記
◆Cajal Advanced Neuroscience Training Programme参加記
◆Participating report of the 12th biennial Conference of CNS : Kelvin Hui
◆「平成29年度 時実利彦記念賞受賞者 齊藤 実先生」
◆「第1回 ジョセフ・アルトマン記念発達神経科学賞受賞者 今吉 格先生」
◆第31回塚原仲晃記念賞受賞者 磯田 昌岐先生
◆第31回塚原仲晃記念賞受賞者 安田 涼平先生
◆小さな脳から探る神経回路
◆大人の脳を維持する仕組み
◆「遺伝学的及び光学的手法による脳内神経活動の可視化と機能解析への応用」
◆「社会」を紐解くニューロサイエンス
◆「海馬への異なる脳領域からの入力の機能と制御」
◆平成29年度 第1回ジョセフ・アルトマン記念発達神経科学賞 受賞者決定
◆第30回塚原仲晃記念賞受賞者 樋口真人先生
◆第30回塚原仲晃記念賞受賞者 坂場武史先生
◆平成28年度時実利彦記念賞受賞者 影山龍一郎先生「神経幹細胞の増殖と分化制御機構の解明とその操作」
◆小型魚類を用いた神経精神疾患研究
◆嗅覚情報処理を支える神経回路の形成基盤
◆視覚から行動へ
◆状況に応じた柔軟な情報処理を可能にする脳神経回路
◆「脳とは何か」の答えを目指して
◆光の動きを検知する視覚神経回路の機能と発達
◆精神疾患病態としてのシナプス病理解明とその治療戦略への展開
◆意識経験はどのような神経メカニズムによって支えられているのか?
◆記憶形成・記憶保持を負に制御する神経回路の研究
◆脳の設計図と変化する脳
◆平成27年度時実利彦記念賞受賞者 渡辺雅彦先生「神経活動依存的な神経回路発達と回路機能発現に関する分子解剖学的研究」
◆第29回塚原仲晃記念賞受賞者 池谷 裕二先生 脳回路活動の構造解析」
◆時差消失マウスの開発による時差の神経分子シグナルの研究
◆サル側頭葉における物体間対連合の視覚表象及び記憶想起を司る神経回路の計算原理
◆てんかんの細胞神経科学
◆嗅覚から記憶へ
◆神経細胞の分化と可塑性を制御するRNAプログラム
◆第28回塚原仲晃記念賞受賞者榎本 和生先生「感覚ニューロン受容野の自己組織化と再編機構の解明」
◆平成26年度時実利彦記念賞受賞者岡本仁先生「脊椎動物脳の進化的保存を利用した情動制御機構の解明」
◆第28回塚原仲晃記念賞受賞者加藤忠史先生 精神疾患の神経生物学的研究」
◆精神医学領域のUnmet medical needsの解消を目指して
◆単純な学習系で照らし出す大脳新皮質に記憶が固定化される仕組み
◆中枢神経疾患における組織傷害と修復を制御する生体応答
◆分子1個1個のふるまいから脳細胞の自己組織化機構を読み解く
◆小脳におけるシナプス形成機構の解明
◆軸索における活動電位の伝導様式とグリア細胞の関与
◆予想に反した結果が発見につながったとき
◆線虫をもちいた温度応答の分子神経メカニズムの解析
◆地図を手に山を登る
◆精神・神経疾患の社会性障害の理解と克服に向けて
◆体液Naレベルのセンシング〜グリア細胞による ニューロンの制御、そして疾患
◆神経科学と心理学の橋渡しを目指して
◆好奇心の原点へ
◆大脳基底核神経回路の制御機構
◆神経軸索ガイダンスの駆動メカニズムの解明を目指して
2021年度時実利彦記念賞受賞者 野田 昌晴 先生
水分および塩分欲求を制御する脳内機構の研究
伝統ある賞にお選びいただき関係者の皆様に感謝申し上げます。
私は大学では、公害問題解決の一助になればと京大工学部の化学系を選んだ訳ですが、修士課程を終える頃には基礎研究の方がおもしろくなり、大学院の後期課程では医学研究科(京大医化学II:沼正作教授)に進学しました。大学院生から助手時代にかけて、エンケファリン前駆体の構造をcDNAクローニングによって明らかにするとともに、ニコチン性アセチルコリン受容体の全サブユニット(α、β、γ、δおよびε)のクローニングを行いました。続いて直ぐに助教授となったため、同教室でさらに数年過ごしてNaチャンネルの構造と機能の研究を行いました。その後、MPI発生生物学研究所(F. Bonhoeffer教授)へ留学しましたが、研究半ばで基礎生物学研究所の教授に選ばれ、急いで帰国することにしました。研究室を立ち上げるに当って3つのテーマを設定しました。①発生期における網膜の領域特異化と視神経の領域特異的投射の機構 ②受容体型プロテインチロシンホスファターゼの生理的役割 ③Naxチャンネルの生理的役割。テーマ①は留学時代の研究を発展させるもの、テーマ③は電位依存的に開口しないために謎のNaチャンネルとして残っていたNaxの役割の解明に挑戦するもので、今回の受賞の対象となったのはこれを発展させたものです。
体液(血液や脳脊髄液を含む細胞外液の総称)のNa+濃度が~145mMに維持される体液恒常性は生命維持にとって必須です。そのために、我々の脳は体液中のNa+濃度を常にモニターしており、その情報に基づいて水分あるいは塩分の摂取と排泄を制御しています。例えば、脱水状態では体液中のNa+濃度が上昇し、ヒトは喉の渇きを覚えて水を飲むとともに、脳下垂体から血中に抗利尿ホルモンが放出され、腎臓での水の再吸収を高め尿量を減少させます。体液恒常性の重要性は早くから認識されていましたが、脳内Naセンサーの実体は長い間不明でありました。
まずNax遺伝子のノックアウト(Nax-KO)マウスを作成し、その発現部位を解析したところ、Naxは脳内の感覚性脳室周囲器官(sCVOs)として知られる脳弓下器官(SFO)と終板脈管器官(OVLT)のグリア細胞(アストロサイトとタニサイト)に発現していました(J. Neurosci. 2000; Am. J. Physiol. 2006)。sCVOsは、脳内で神経細胞が存在しているにもかかわらず血液-脳関門が欠失している部位であり、第3脳室に面していることから、血液と脳脊髄液の両方の状態をモニターするのに適した場所でした。Naxを発現する細胞で細胞外液のNa+濃度を徐々に上げていくと、生理的濃度を越えた~150mMで開口し始めNa+流入が観察されました(Nat. Neurosci. 2002)。さらに、このNaxの活性化の[Na+]の閾値は、エンドセリン-3(ET-3)によって低濃度側にシフトすること、脱水条件下ではSFOにおいてET-3の発現が始まることを発見しました(Cell Metab. 2013)。脱水状態では、野生型のマウスは塩分摂取を抑制しますが、Nax-KOはこれを抑制せず、あたかもNaセンサーが欠失しているような行動を示します(J. Neurosci. 2000)。また、Naxに対する自己免疫疾患の患者は「口渇感を伴わない高Na血症」を発症していました(Neuron 2010)。
その後、SFOのNaxシグナルは主に塩分摂取の抑制(J. Neurosci. 2004)に、OVLTのNaxシグナルは水分摂取の誘導(Am. J. Physiol. 2016)に働いていることを突きとめました。Naxはグリア細胞に発現していますが、SFOでは、Naxの活性化の結果グリア細胞から分泌された乳酸が近傍の抑制性ニューロン(GABAニューロン)を活性化することによって、塩分摂取誘導ニューロンの活性を抑制し(Neuron 2007)、またOVLTでは、Naxの活性化によってグリア細胞からepoxyeicosatrienoic acids(EETs)が分泌され、近傍のTRPV4を発現する水分摂取誘導ニューロンを活性化していました(Am. J. Physiol. 2016)。
出血等で体液を失ったときには、水分と塩分の両方に対して渇望が生じますが、これは血液中のアンジオテンシンⅡ(AngⅡ)が上昇するためです。SFO内にAngⅡ受容体(AT1a)を発現する細胞群があり、これらが、OVLTあるいは腹側分界条床核(vBNST)に連絡していることを見出しました(Nat. Neurosci. 2017; 図1)。前者(水ニューロン)は水分欠乏時に、後者(塩ニューロン)は塩分欠乏時に活性化します。光遺伝学の手法を用いて、人為的に前者を活性化すると水分摂取が誘導され、後者を活性化すると塩分摂取が誘導されます。また、脳室内のコレシストキニン(CCK)は水分摂取を抑制しますが、塩分欠乏時にはSFO内でCCKの発現上昇が起こることを発見しました。このCCKはSFO内のCCK作動性ニューロンで作られており、CCK-B受容体陽性のGABAニューロンの活性化を介して水分摂取(水ニューロン)を抑制していること、さらに、CCKニューロンには、塩欠乏時に持続的に活性化するものと、飲水に伴って一過性に活性化するものの2種類があることが明らかになりました(Nat. Commun. 2020; 図2)。光遺伝学を用いて、それぞれのCCKニューロンを抑制すると水分摂取が誘導されることから、いずれも水分の過剰摂取を抑制する役割であることが分かります。最近、OVLTでは、Naxに加えて、SLC9A4(Na+/H+対向輸送体)も独立して体液のNa+濃度上昇を検知しており、両者が共力して水分摂取を制御していることが判明しました(Eur. J. Phys. 2020)。
塩分の過剰摂取によって体液のNa+濃度が上昇すると高血圧を発生しますが、Nax-KOマウスでは、体液のNa+濃度は野生型と同様に上昇するものの、高血圧を発症しません。そのシグナル伝達機構として、OVLTのNaxが[Na+]上昇を感知して活性化し、その結果Naxを発現するグリア細胞から乳酸とともにH+が分泌され、近傍の酸感受性チャンネル(ASIC1a)陽性のニューロンを活性化し、下流の交感神経制御中枢(室傍核(PVN)および頭側延髄腹外側野(RVLM))の活性化を通して、血圧上昇を誘発していることが分かりました(Neuron 2018)。
今後は、塩分摂取に伴う抑制メカニズム、並びに神経性高血圧の発生機序について研究できたらと思っております。
私は大学では、公害問題解決の一助になればと京大工学部の化学系を選んだ訳ですが、修士課程を終える頃には基礎研究の方がおもしろくなり、大学院の後期課程では医学研究科(京大医化学II:沼正作教授)に進学しました。大学院生から助手時代にかけて、エンケファリン前駆体の構造をcDNAクローニングによって明らかにするとともに、ニコチン性アセチルコリン受容体の全サブユニット(α、β、γ、δおよびε)のクローニングを行いました。続いて直ぐに助教授となったため、同教室でさらに数年過ごしてNaチャンネルの構造と機能の研究を行いました。その後、MPI発生生物学研究所(F. Bonhoeffer教授)へ留学しましたが、研究半ばで基礎生物学研究所の教授に選ばれ、急いで帰国することにしました。研究室を立ち上げるに当って3つのテーマを設定しました。①発生期における網膜の領域特異化と視神経の領域特異的投射の機構 ②受容体型プロテインチロシンホスファターゼの生理的役割 ③Naxチャンネルの生理的役割。テーマ①は留学時代の研究を発展させるもの、テーマ③は電位依存的に開口しないために謎のNaチャンネルとして残っていたNaxの役割の解明に挑戦するもので、今回の受賞の対象となったのはこれを発展させたものです。
体液(血液や脳脊髄液を含む細胞外液の総称)のNa+濃度が~145mMに維持される体液恒常性は生命維持にとって必須です。そのために、我々の脳は体液中のNa+濃度を常にモニターしており、その情報に基づいて水分あるいは塩分の摂取と排泄を制御しています。例えば、脱水状態では体液中のNa+濃度が上昇し、ヒトは喉の渇きを覚えて水を飲むとともに、脳下垂体から血中に抗利尿ホルモンが放出され、腎臓での水の再吸収を高め尿量を減少させます。体液恒常性の重要性は早くから認識されていましたが、脳内Naセンサーの実体は長い間不明でありました。
まずNax遺伝子のノックアウト(Nax-KO)マウスを作成し、その発現部位を解析したところ、Naxは脳内の感覚性脳室周囲器官(sCVOs)として知られる脳弓下器官(SFO)と終板脈管器官(OVLT)のグリア細胞(アストロサイトとタニサイト)に発現していました(J. Neurosci. 2000; Am. J. Physiol. 2006)。sCVOsは、脳内で神経細胞が存在しているにもかかわらず血液-脳関門が欠失している部位であり、第3脳室に面していることから、血液と脳脊髄液の両方の状態をモニターするのに適した場所でした。Naxを発現する細胞で細胞外液のNa+濃度を徐々に上げていくと、生理的濃度を越えた~150mMで開口し始めNa+流入が観察されました(Nat. Neurosci. 2002)。さらに、このNaxの活性化の[Na+]の閾値は、エンドセリン-3(ET-3)によって低濃度側にシフトすること、脱水条件下ではSFOにおいてET-3の発現が始まることを発見しました(Cell Metab. 2013)。脱水状態では、野生型のマウスは塩分摂取を抑制しますが、Nax-KOはこれを抑制せず、あたかもNaセンサーが欠失しているような行動を示します(J. Neurosci. 2000)。また、Naxに対する自己免疫疾患の患者は「口渇感を伴わない高Na血症」を発症していました(Neuron 2010)。
その後、SFOのNaxシグナルは主に塩分摂取の抑制(J. Neurosci. 2004)に、OVLTのNaxシグナルは水分摂取の誘導(Am. J. Physiol. 2016)に働いていることを突きとめました。Naxはグリア細胞に発現していますが、SFOでは、Naxの活性化の結果グリア細胞から分泌された乳酸が近傍の抑制性ニューロン(GABAニューロン)を活性化することによって、塩分摂取誘導ニューロンの活性を抑制し(Neuron 2007)、またOVLTでは、Naxの活性化によってグリア細胞からepoxyeicosatrienoic acids(EETs)が分泌され、近傍のTRPV4を発現する水分摂取誘導ニューロンを活性化していました(Am. J. Physiol. 2016)。
出血等で体液を失ったときには、水分と塩分の両方に対して渇望が生じますが、これは血液中のアンジオテンシンⅡ(AngⅡ)が上昇するためです。SFO内にAngⅡ受容体(AT1a)を発現する細胞群があり、これらが、OVLTあるいは腹側分界条床核(vBNST)に連絡していることを見出しました(Nat. Neurosci. 2017; 図1)。前者(水ニューロン)は水分欠乏時に、後者(塩ニューロン)は塩分欠乏時に活性化します。光遺伝学の手法を用いて、人為的に前者を活性化すると水分摂取が誘導され、後者を活性化すると塩分摂取が誘導されます。また、脳室内のコレシストキニン(CCK)は水分摂取を抑制しますが、塩分欠乏時にはSFO内でCCKの発現上昇が起こることを発見しました。このCCKはSFO内のCCK作動性ニューロンで作られており、CCK-B受容体陽性のGABAニューロンの活性化を介して水分摂取(水ニューロン)を抑制していること、さらに、CCKニューロンには、塩欠乏時に持続的に活性化するものと、飲水に伴って一過性に活性化するものの2種類があることが明らかになりました(Nat. Commun. 2020; 図2)。光遺伝学を用いて、それぞれのCCKニューロンを抑制すると水分摂取が誘導されることから、いずれも水分の過剰摂取を抑制する役割であることが分かります。最近、OVLTでは、Naxに加えて、SLC9A4(Na+/H+対向輸送体)も独立して体液のNa+濃度上昇を検知しており、両者が共力して水分摂取を制御していることが判明しました(Eur. J. Phys. 2020)。
塩分の過剰摂取によって体液のNa+濃度が上昇すると高血圧を発生しますが、Nax-KOマウスでは、体液のNa+濃度は野生型と同様に上昇するものの、高血圧を発症しません。そのシグナル伝達機構として、OVLTのNaxが[Na+]上昇を感知して活性化し、その結果Naxを発現するグリア細胞から乳酸とともにH+が分泌され、近傍の酸感受性チャンネル(ASIC1a)陽性のニューロンを活性化し、下流の交感神経制御中枢(室傍核(PVN)および頭側延髄腹外側野(RVLM))の活性化を通して、血圧上昇を誘発していることが分かりました(Neuron 2018)。
今後は、塩分摂取に伴う抑制メカニズム、並びに神経性高血圧の発生機序について研究できたらと思っております。
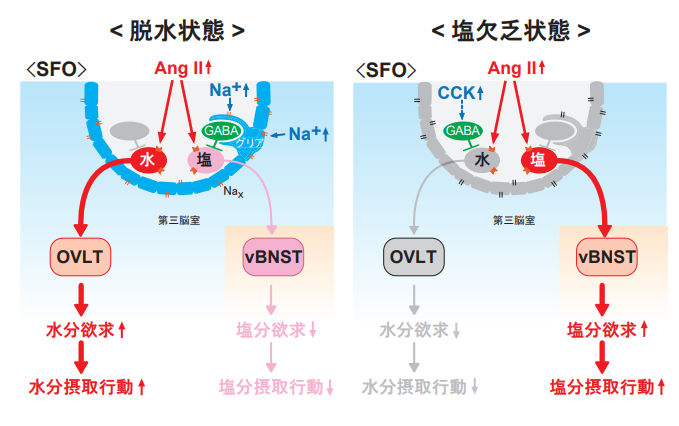
図1. 水分欲求および塩分欲求の制御機構
脱水状態(左)では、AngⅡとNa+の両方の濃度が高まる。AngⅡは水ニューロンと塩ニューロンの両方に作用するが、Na+濃度上昇によってNax陽性のグリア細胞が活性化し、続いてGABAニューロンが活性化するため、塩ニューロンの活動は抑えられ、専ら水ニューロンが活性化する。塩欠乏状態(右)では、AngⅡとCCKの両方の濃度が高まる。CCKが上記とは別のGABAニューロンを活性化することにより、水ニューロンの活動は抑えられ、塩ニューロンだけが活性化する。
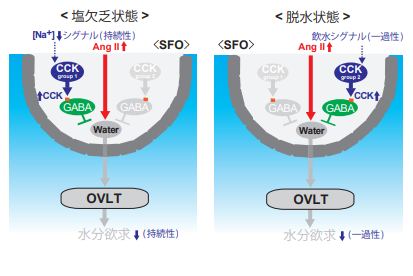
図2. 水分欲求が抑制される仕組みの詳細
塩欠乏状態(左)ではAngⅡ濃度は上昇しているが、体液中のNa⁺濃度は低下しており、同条件下で持続的に活性化状態にあるグループ1のCCKニューロンによって水分摂取は抑制される。また脱水状態(右)では水ニューロンは通常活性化状態にあるが、その活動は飲水に反応してそのつど一過性に抑制され、水分欲求は一時的に収まる。これはグループ2のCCKニューロンの働きによる。水ニューロンは、このように情況に応じてSFO内の異なるCCKニューロンによって抑制制御を受ける。

野田 昌晴(東京工業大学・科学技術創成研究院)
略歴
1977年3月 京都大学工学部工業化学科 卒業
1979年3月 京都大学工学研究科修士課程工業化学専攻 修了
1983年10月 京都大学医学研究科博士課程生理系専攻 修了
1983年4月 日本学術振興会 奨励研究員
1984年4月 京都大学医学部 助手
1985年4月 京都大学医学部 助教授
1989年4月-1991年8月 マックスプランク発生生物学研究所 客員研究員
1991年9月 基礎生物学研究所 教授、総合研究大学院大学 教授
2019年4月 東京工業大学 科学技術創成研究院 特任教授
1977年3月 京都大学工学部工業化学科 卒業
1979年3月 京都大学工学研究科修士課程工業化学専攻 修了
1983年10月 京都大学医学研究科博士課程生理系専攻 修了
1983年4月 日本学術振興会 奨励研究員
1984年4月 京都大学医学部 助手
1985年4月 京都大学医学部 助教授
1989年4月-1991年8月 マックスプランク発生生物学研究所 客員研究員
1991年9月 基礎生物学研究所 教授、総合研究大学院大学 教授
2019年4月 東京工業大学 科学技術創成研究院 特任教授



HOME | 一般社団法人 日本神経科学学会
- 学会機関紙
- Neuroscience Research
- Articles in Press
- Latest Issue
- Back Issues
- Submit Your Paper
- About the Journal
- 電子版の購読について
(会員の方へ) - お知らせ
- 公募情報 助成・受賞
- 研究員・教職員募集
- 大学院生募集・説明会
- 研究助成・渡航助成募集
- 受賞候補者募集
- キャリアパスに応じた賞・助成
- 海外の学会への旅費支援
- 学会推薦による賞・助成
- 成茂神経科学研究助成基金
- イベント・研究会情報
- イベント・研究会
- 一般の方向けイベント
- 神経科学の発展のために
- 倫理問題などに関する指針
- 動物実験に関する指針
- MRI検査の指針
- ランチョン大討論会
- ダイバーシティの取り組みについて
- 会員ページ
- Neuroscience Research (NSR) 電子版閲覧
- 抄録検索システム [IDパスワードが必要]
- 会員ログイン
- 過去の神経科学ニュース